In today’s research landscape, a cell biologist faces into two seemingly endless chasms of content in their regular study of disease understanding and novel therapy development. First, a scientist must address the tremendous amount of knowledge revealed in new molecular biology/genetics publications, and second, the sheer number of data points generated by OMICs technologies for a single experiment. The second challenge in fact compounds the first as, without programming or advanced bioinformatics skills, many cell biologists lack the tools to interpret their own experimental data.
Despite the fact that specialized cells in a multicellular organism have characteristic patterns of gene expression, each cell is capable of altering its pattern in response to extracellular cues. For example, if a liver cell is exposed to a glucocorticoid hormone, protein production dramatically increases, as it would in order to elevate energy production during starvation or exercise. When the hormone is no longer present, production of these proteins drops to its normal level. Other cell types respond to glucocorticoids differently, and some do not respond at all. Although this example is somewhat oversimplified, it illustrates the general feature of cell specialization wherein lays the basis of many diseases caused by system modulation by external factors, such as environmental or genetic influence.
Normally, cells respond to extracellular signals by initiation of molecular chain reaction cascades known as pathways, which may cause changes in numerous cell characteristics including metabolism activation, division, growth, cytoskeleton remodeling and more depending on a cell’s specialization. These pathways connect signal receptors, lead (through a number of adaptor proteins or second messengers) to activation/inhibition of proteins presented in a cell, and may eventually affect gene expression pattern.
This process is widely used as the basis for disease and novel targeted therapy research as well as for existing drugs repositioning. Through analysis of disease related gene expression patterns generated by high throughput techniques, one can construct a global picture of significantly over- or under-expressed genes in disease tissue samples compared to control samples (also called differentially expressed genes or DEGs). By identifying these alterations’ place on a known map of cellular processes, one may interpret the biological effect of these molecular changes.
As noted previously, this pathway analysis approach is not a simple process due to the size of the gene set, and is certainly not a task that could be done manually. Researchers therefore require automated approaches to understanding the overlaps of OMICs data sets with existing schemas of cellular pathways, in order to highlight cellular processes changes on a system level. This requirement has created a market for not only skilled bioinformaticians, but also software to simplify the process. The success possible through support from bioinformaticians and/ or software solutions in reducing the complexity of complex biological systems has made systems biology and pathway approaches a widely popular and accepted solution to the content chasm that cellular biologists stare into at the start of a research project.
Despite being very popular, methods supporting pathway analysis have certain limitations and two questions should be considered when approaching them:
1. Does the existing pathway knowledge cover all genes from my list?
2. How can I distinguish cause and effect?
Question 1: Does the existing pathway knowledge cover all genes from my list?
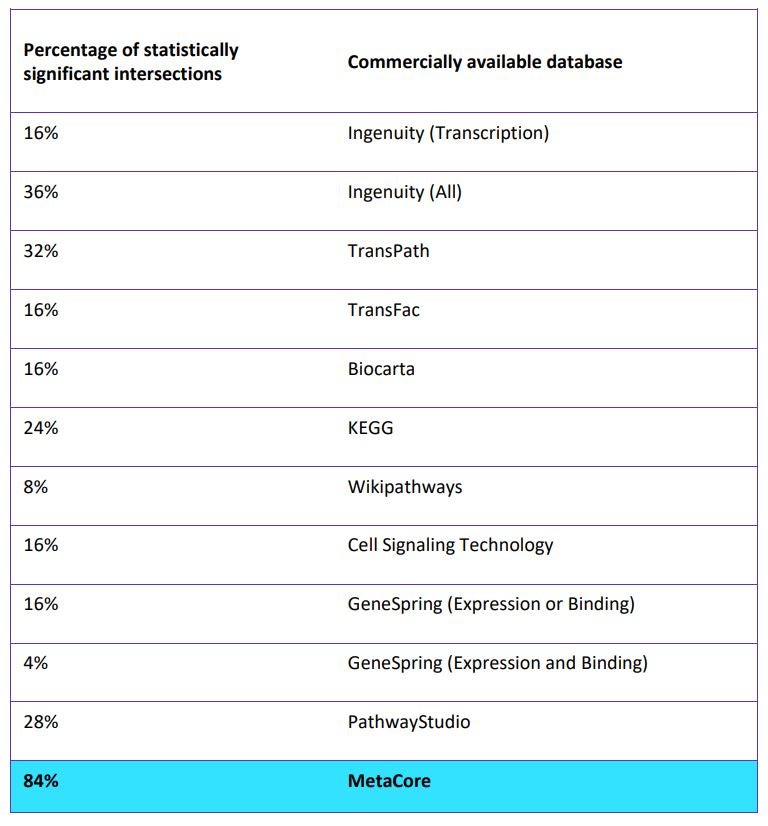
Existing knowledge about pathways and cellular processes is not universal, so the number of proteins contained in pathway databases differ and do not cover the whole genome, just overlaying DEGs on pathways. Therefore, content from one database alone may not be sufficient to ensure complete disease mechanism understanding. A study conducted by the Center for Health Informatics and Bioinformatics, New York University School of Medicine, identified that the overlap between experimentally obtained target genes and targets reported in transcriptional regulatory pathway databases is surprisingly small and often is not statistically significant[1]. The only database that yielded statistically significant overlap was MetaCore, a Cortellis solution.
Question 2: How can I distinguish cause and effect?
This question is critical. Do the DEGs affect cellular processes and pathway activity leading to disease phenotype, or are DEGs results of aberrant signaling pathway activity themselves? In simple terms, are DEGs the cause or effect of the disease phenotype? Most likely both processes are involved in disease progression on the molecular level, but capturing the causation vs. correlation is a challenge that pathway enrichment alone cannot complete.
Endeavoring to answer these two questions leads us to a realm beyond simply the canonical pathway maps available.
Gene expression is orchestrated by proteins called “transcriptional factors”. While some transcriptional factors are constitutively active to maintain cell functions, others may be activated by a signaling pathway. However, all activate or suppress transcription of genes. Transcription factors do not just regulate a single gene, but in fact serve as “master switches” for multiple genes to facilitate gene expression changes in response to stimuli.
One who has knowledge of which gene’s expression is activated/suppressed by which transcription factor can reconstruct a pattern of activated and inhibited transcription factors that stand behind DEG lists obtained in OMICs experiments. Using that transcription factor pattern, one can then continue by using signaling cascade understanding to identify kinases and adaptor proteins activity changes that may be responsible for transcription factor activity alterations. This powerful method is called “reverse causal reasoning” and it allows inference of molecular activity changes which are concordant with observed gene expression but could not be captured by transcriptomics techniques alone (Figure 1). It is possible to identify key hubs connecting several affected transcription factors. Just like a puppeteer manipulates puppets by touching strings, the key hub manipulates gene expression by activating and inhibiting transcriptional factors or by affecting gene expression directly.
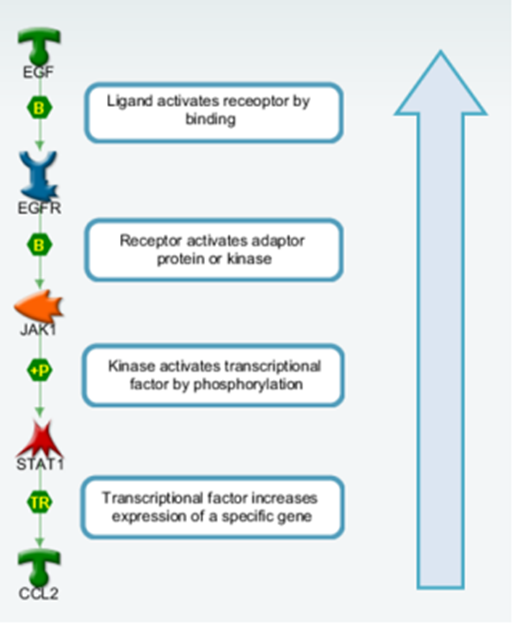
Figure 1: A schematic visualization of a pathway that increases expression of one gene. The reverse causal reasoning logic will start analysis from the bottom. If CCL2 molecule has increased expression the algorithm interprets it as potential increased activity of STAT1 transcriptional factor. If other genes’ expression positively regulated by STAT1 is also elevated in the dataset (and conversely the genes suppressed by STAT1 have decreased expression), then the algorithm goes one step upstream to find the most probable activator of STAT1. On the other hand, if CCL2 has decreased expression so as other genes positively regulated by STAT1, then the algorithm interprets that as STAT1 is in a predominantly inactive state and looks for potential inhibitors upstream that would match this alongside other observed transcription factors in the dataset as a potential “causal” node.
Having such a list of expression and predicted activity changes, a scientist can then investigate whether or not similar changes are known for similar conditions and identify agreement with prior knowledge. Such understanding enables the possibility of finding novel gene behavior in relation to the disorder. Determining if some molecules are known targets for drug development in similar diseases also helps to identify molecules that may be disease causing.
Collection of potential causal activity changes may help in the identification of the most probable disease causing mechanisms and responsible genes. Such genes are a natural opportunity for further drug development or repositioning efforts. However, data collection for such analysis can be a significant task due to the need for a large and complete database of molecular interactions and disease relationships. Such database construction requires analysis of millions of scientific publications with sufficient scientific rigor to ensure that each relationship is reliable enough to make decisions upon.
The current trend in using free public databases unfortunately does not solve this problem. Public databases are reliant upon the curation efforts of a community or individual team and since the information is “free,” the level of pressure to ensure high quality information is reduced. These databases often also lack the basic content required to correctly perform analysis. For example, most public databases lack interaction effect (activation or inhibition), which alone is enough to make causal reasoning approaches impossible. Couple this with the fact that transcription factor action on regulated genes are scarcely annotated; biomarker databases predominantly cover clinically approved data, rather than the required early research expression changes; and public pathway databases contain limited numbers of genes associated with processes. Unfortunately, the utility of public data, even when combined, is a highly dubious.
Find out more about how Clarivate can help accelerate your drug discovery research.
References:
[1] Shmelkov E, Tang Z, Aifantis I, Statnikov A. Assessing quality and completeness of human transcriptional regulatory pathways on a genome-wide scale. Biol Direct. 2011 Feb 28;6:15. doi: 10.1186/1745-6150-6-15. PMID: 21356087; PMCID: PMC3055855.