What are the most important factors to consider when forecasting the launch of generic Xeljanz?
Pfizer’s Xeljanz was the first Janus kinase (Jak) inhibitor to launch in a major market, after its approval as a treatment for rheumatoid arthritis (RA) in the United States in 2012. It is now approved in all major markets for multiple indications (Table 1). In addition to RA, psoriatic arthritis (PsA), and ulcerative colitis (UC), we expect Xeljanz to launch as a treatment for axial spondyloarthritis (AxSpA) in the United States and Europe by 2022. As we prepare to forecast our expectations for these markets from 2017 to 2027, we are considering how Xeljanz’s patent expiry and expected generic launch will shape market dynamics in the next ten years.
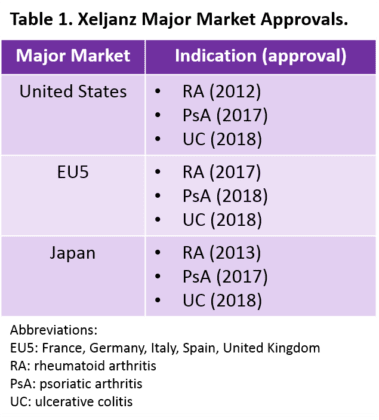
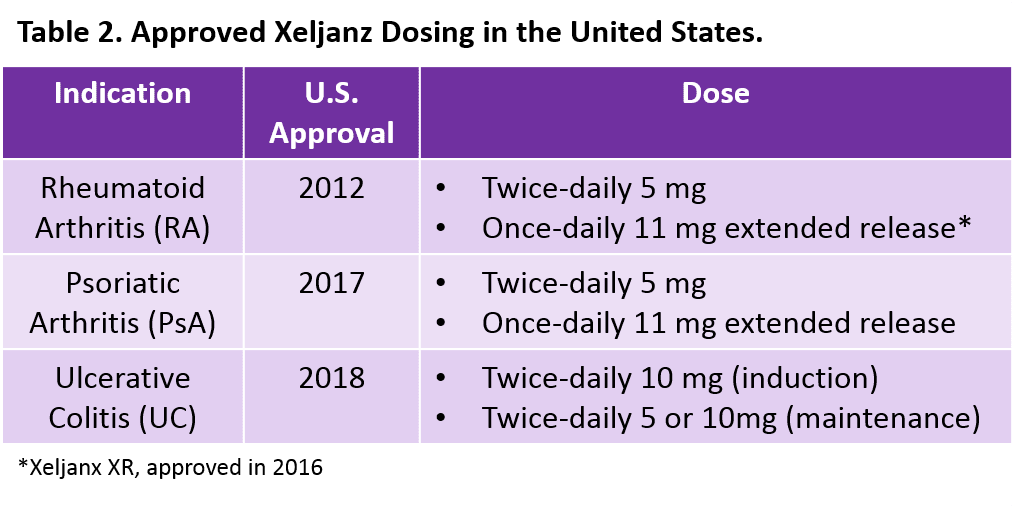
Xeljanz is available in both an immediate-release formulation with two different twice-daily doses and an extended-release (Xeljanz XR) formulation with once-daily dosing (Table 2). However, to date, Xeljanz XR is only available in the United States (and is only approved for RA and PsA). According to the FDA’s Orange Book, one of the strongest patents for Xeljanz expires in December 2025.1 Even if Xeljanz receives a potential pediatric extension for juvenile idiopathic arthritis2,3 giving it six additional months of patent protection, the twice-daily formulation is expected to face generic competition within the next 10 years. However, we expect that once-daily Xeljanz XR will remain under patent beyond our 10-year forecast period.1 Our research also suggests that twice-daily Xeljanz could face generic competition in Japan by 2026 and in Europe by 2027.4,
To inform our forecasts, we have spoken with—and will continue conversations with—colleagues in other therapy areas at DRG to find potential market analogues that will help in guiding our assumptions. Below we outline some potential issues that will likely impact uptake of generic Xeljanz:
- Availability of multiple forms: With Xeljanz and Xeljanz XR available for RA and PsA, we are using our primary market research to understand if physicians are choosing to switch from one formulation to another. All other Jak inhibitors approved (Eli Lilly’s Olumiant [baricitinib]) or in development (AbbVie’s upadacitinib, Gilead/Galapagos’s filgotinib) for these indications benefit from a once-daily dosing profile. Therefore, Pfizer is likely to use an aggressive switch campaign to move patients from the twice-daily formulation facing patent loss to the XR version, which offers competitive once-daily dosing and extended patent protection. Although Pfizer may seek approval of once-daily Xeljanz XR in Europe (and has a patent application for this formulation pending), no formal announcement has been made to date. Xeljanz XR is not approved in Japan either, but a Phase III clinical trial with sites in Japan and a primary completion date of March 2017 is registered in ClinicalTrials.gov, suggesting the possibility of approval.6 One possible analogue we are considering as a market model for Xeljanz/Xeljanz XR is the antidepressant Effexor/Effexor XR; also marketed by Pfizer, Effexor experienced patent loss before the XR version.
- Efficacy and safety dynamics within the drug class: As the first Jak inhibitor available in a major market, Xeljanz was the only available oral targeted therapy in the United States for RA from 2012 to 2018 (when Olumiant was approved). However, unlike Xeljanz, which targets Jak-1 and Jak-3 (or even Olumiant, which targets Jak-1 and Jak-2), the newest Jak inhibitors in development, upadacitinib and filgotinib, target Jak-1 only. Some interviewed physicians speculate that these agents, with increased Jak specificity, may prove to have better efficacy and a more favorable safety profile as compared with older Jaks. For example, one German rheumatologist stated:
“Filgotinib and upadacitinib are in clinical trials right now and because they are relatively selective for Jak-1 it will be interesting to see how the side effect profile is in comparison to Xeljanz and baricitinib.”
Sufficient differentiation of these newer Jak inhibitors could limit the market impact of generic Xeljanz. A possible market analogue for these drug class effects may be the entry of statins into the cardiovascular space where newer, improved branded agents competed against older, generically-available therapies
- Intra and inter-class effects: As the first oral targeted therapy approved for RA, PsA, and UC, Xeljanz is a welcome addition to physicians’ armamentarium. In RA, Xeljanz is generally used as a later line therapy due to physician comfort with older targeted therapies and payer restrictions that favor more-established agents; a similar trend is expected for uptake in PsA and UC. As physicians and patients become more comfortable with Jak inhibitors, and especially with their safety profiles, the entry of a generic Jak may affect the patient share of not only those agents within the same class, but also of other available therapies, especially those without a biosimilar alternative or those used later in the treatment algorithm. A possible analogue we are considering to better understand generic Xeljanz’s impact on the future treatment algorithm for RA, PsA, UC, and AxSpA is Novartis’s Gilenya, indicated for MS, and the launch assumptions our colleagues in CNS developed for its generic entry (although a recent patent win is now expected to delay its generic entry until after Xeljanz).7 Gilenya, like Xeljanz, was the first oral therapy available for MS and has a position in the MS treatment algorithm comparable to Xeljanz’s in RA, PsA, and UC. In addition, novel agents in the same class as Gilenya are in late stage clinical trials, analogous to Jak inhibitors in development that are expected to compete with Xeljanz.
- Regional variability: Differences between the individual markets we cover (United States, EU5, and Japan) including the timing of generic Xeljanz entry, which is expected to be staggered due to region-specific patent and exclusivity periods, will impact our markets assumptions. The United States and Japan will most likely be the first major markets with a Xeljanz generic, in 2026. A generic in the EU5 will follow as early as 2027, although additional market protections could occur if Xeljanz launches in a new indication where it brings significant clinical benefit over existing therapies. A possible market analogue to model the complex dynamics of regional variability with respect to patent expiry is Eli Lilly’s Cymbalta, first marketed in the United States for depression in 2004 and subsequently approved for neuropathic pain, generalized anxiety disorder, fibromyalgia, and chronic musculoskeletal pain. Generics of Cymbalta were first available in the United States at the end of 2013 followed by Europe at the beginning of 2015, a similar timeline to our expectations for Xeljanz.
- Price: Of course, in an increasingly cost-conscious healthcare environment, price is a major factor when predicting market dynamics of a new generic drug. Currently the price of branded Xeljanz is similar to (and often lower than) prices of targeted therapies used in the RA, PsA, and UC markets. Therefore, we believe that it is unlikely that generic manufacturers will launch their drugs with a price that is pennies on the dollar. Nevertheless, as a small molecule therapy, we expect easier barriers to generic entry compared with biosimilar development, leading to an increased number of generic competitors. As such, generic Xeljanz will likely be deeply discounted relative to the brand and the biosimilars with which it is expected to compete. A competitively priced generic would encourage use, especially by price conscious European healthcare systems. A potential model for pricing may be the migraine market where generic triptans can have different discounts relative to their branded agent; several factors, including the method of administration and the number of generic competitors, can impact the extent of the discount.
It remains to be seen how these markets (RA, PsA, UC, and AxSpA) will respond to the launch of generic Xeljanz and although no single analogue mentioned above is a perfect match for the specific dynamics of the Jak inhibitor class, we are taking advantage of DRG’s subject area experts’ extensive knowledge of related markets to help us determine how we should forecast the emergence of generic Xeljanz.
For more detail, see our Disease Landscape and Forecast report for your indication of interest.
References:
- https://www.accessdata.fda.gov/scripts/cder/ob/. Accessed 7 August 2018.
- Approved Active Moieties to which FDA has issued a Written Request for Pediatric Studies under Section 505A of the Federal Food, Drug, and Cosmetic Act. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm050002.htm. Accessed 7 August 2018.
- Clinical Trials.gov. JIA studies registered with tofacitinib. https://clinicaltrials.gov/ct2/resultsterm=tofacitinib&cond=%22Arthritis%22&age_v=&age=0&gndr=&type=&rslt=&Search=Apply. NCT01513902, NCT01932372, NCT01500551, NCT02592434, NCT03000439. Accessed 7 August 2018.
- Ark Patent Intelligence 2018. https://www.arkpatentintelligence.com/login
- Information on Japanese Regulatory Affairs. Accessed 27 August 2018. http://www.jpma.or.jp/english/parj/pdf/2018.pdf
- A Study To Evaluate The Safety And Efficacy Of Tofacitinib Modified Release Tablets Compared To Tofacitinib Immediate Release Tablets In Adult Patients With Rheumatoid Arthritis. https://clinicaltrials.gov/ct2/show/NCT02281552. Accessed 27 August 2018.
- Gilenya patent decision gives MS market a reprieve. Biopharma Dive https://www.biopharmadive.com/news/gilenya-patent-decision-gives-ms-market-a-reprieve/527758/. Accessed 7 August 2018.
We’d like to thank DRG’s Central Nervous System and Cardiovascular Teams for in depth discussions of their markets as well as Ajay Puri and the Japanese team for their assistance in performing patent research.




Related insights
The latest news, technologies, and resources from our team.