第七屆中國罕見病高峰論壇於2018年9月14-16日在上海舉辦,此論壇的目的在於增加對於罕見病的理解和認識。在此之前,科睿唯安生命科學解決方案顧問 趙宇薇 於8月初的 webinar中對罕見病及罕見病藥物(也指孤兒藥,即orphan drug)在中國的發展現況進行討論,本文節錄該次webinar內容,且特別感謝科睿唯安生命科學資深編輯 Shyama Ghosh女士共同完成本文。
全球主要藥政市場罕見病藥物政策
在今年(2018)的5月22日,中國公布了第一批罕見病目錄,其中一共列出121個罕見病;此舉被視為中國政府重視罕見病及相應罕見病藥物市場需求的第一步。如圖1所示,在中國目前可經由特殊審批的快速通道加速罕見病藥上市 (參考2017年126號文),其他主要的藥政市場如美國、歐盟及澳洲等,也有相應推動新藥,特別是罕見病藥物上市的特殊審查及加速機制。
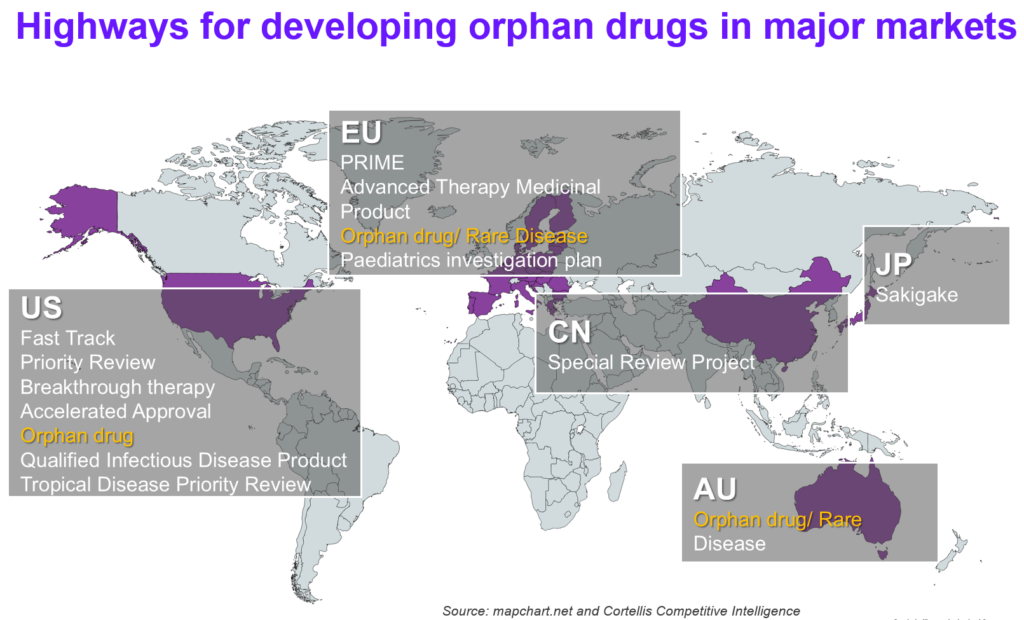
表1列舉了美國、歐洲及日本三個主要市場發展罕見病藥物的法規及鼓勵措施。
| 國家 | 美國 | 歐盟 | 日本 |
|---|---|---|---|
| 法源基礎 | 21 CFR part 316
孤兒藥法案 |
Regulation (EC) No 141/2000 | 藥事法第77-2條 |
| 主管機關 | FDA;
孤兒藥產品開發辦公室 (Office of Orphan Products & Development) |
EMA;
孤兒用藥委員會 (Committee for Orphan Medicinal Products, COMP) |
厚生勞動省 (MHLW);
藥事食品衛生省議會 (Pharmaceutical Affairs & Food Sanitation Council ) |
| 罕見病定義 | 少於20萬人 | 低於1/2000(約少於25萬人) | 少於5萬人 |
| 政策獎勵 |
|
|
|
罕見病藥物在美國FDA的獲批趨勢
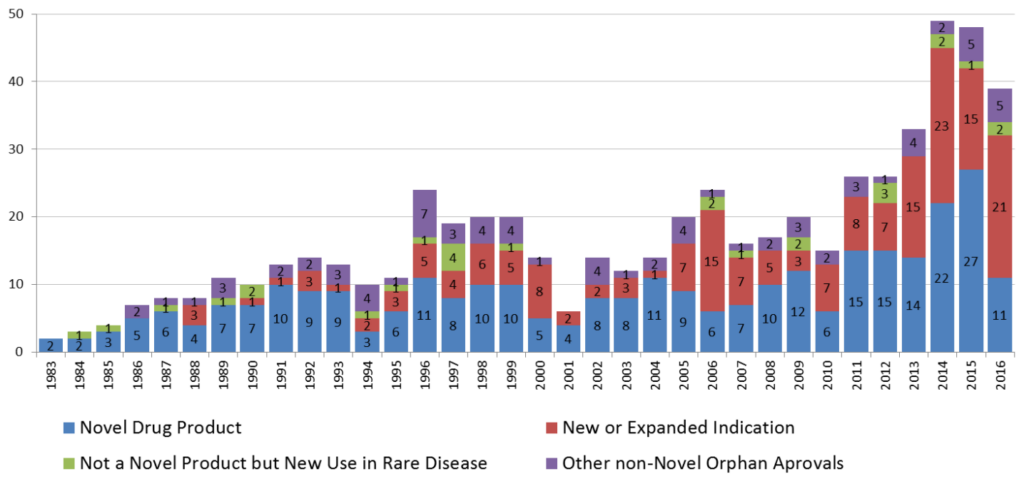
罕見病或許比您我想像的更普遍,根據統計,目前約有 7000 種罕見病1,全球罹患罕見病的人口約有3億人,80%的罕見病和基因有關,而50%的罕見病患者是兒童。美國FDA在1983年頒布了孤兒藥法案後,迄今約有450個罕見病藥物獲准上市,其中部分的藥物是透過取得罕見病藥物資格上市,而有部分藥物則是以適應症拓展等方式獲得罕見病藥物資格。圖2是FDA公告1983年以來罕見病藥物獲准取得上市許可的統計結果,根據FDA的分析,2017年共有77個罕見病藥物獲准上市,上市成功率達100%,涵蓋腫瘤 (58%)、其他 (10%)及代謝疾病 (7%)、神經疾病 (7%) 等主要治療領域。然而,即使在高獲准率的情勢之下,仍有95%的罕見病無藥可治2。
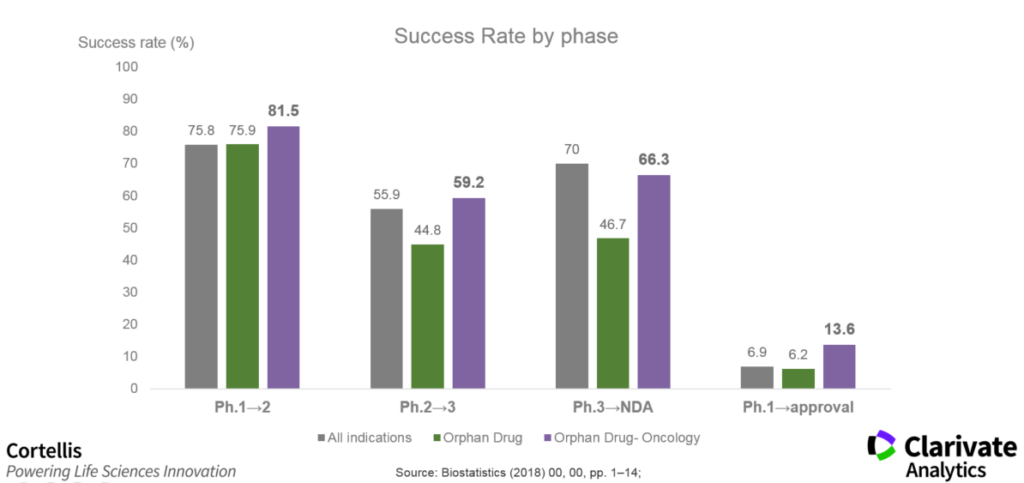
罕見病藥物的臨床成功率
既然如FDA公布的在2017年罕見病藥物獲批的成功率是100%,那麽在臨床試驗上的成功率呢?從2018年Wong et al.的研究指出,從臨床一期起算到取得上市許可,罕見病藥物平均的成功率是6.2%,略低於其他適應症用藥的成功率(6.9%)。若把腫瘤相關的適應症排除後,罕見病藥物(不含腫瘤用藥)的成功率則增加至13.6% (見圖3)。
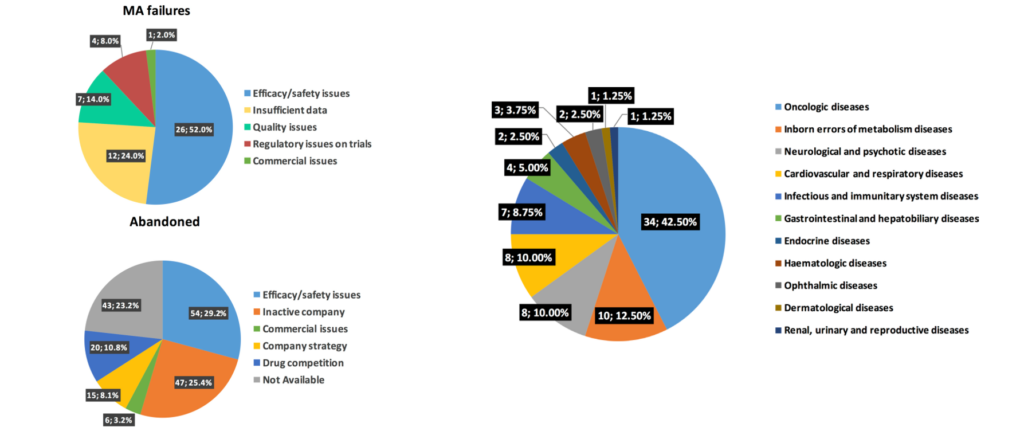
罕見病藥物失敗原因分析
圖4描繪了在歐洲罕見病藥物未能取得上市許可,或在研發過程中被放棄導至中斷的原因(圖4左),以及進一步分析因安全性/有效性失敗的罕見病藥物的治療領域(圖4右)。與其他在研發過程中失敗的藥物相似,罕見病藥物失敗的主要原因還是在安全性/有效性(efficacy/safety issue)。在未能取得藥證的罕見病藥物當中,前三大的失敗原因分別是:安全性/有效性(52%)、數據不充分(insufficient data) (24%)以及品質(quality issue)(14%);50%的失敗是發生在臨床二期。進一步分析因安全性/有效性失敗的罕見病藥物的治療領域,分別是腫瘤、 遺傳性代謝疾病(inborn errors of metabolism diseases)、神經及精神疾病、以及心血管及呼吸相關疾病。
研發成本及罕見病治療成本
罕見病藥物與非罕見病藥物的研發成本差距在於臨床試驗,如表2所示,罕見病藥物的臨床試驗成本在扣除稅務抵减前是非罕見病藥物的50% 3;Linda Martin的研究指出,執行非罕見病藥物全球臨床試驗的成本中位數約是3,340萬美元4,因此,若考慮臨床試驗成本及稅務抵减,罕見病藥物的臨床試驗成本中位數約是在835萬美元。
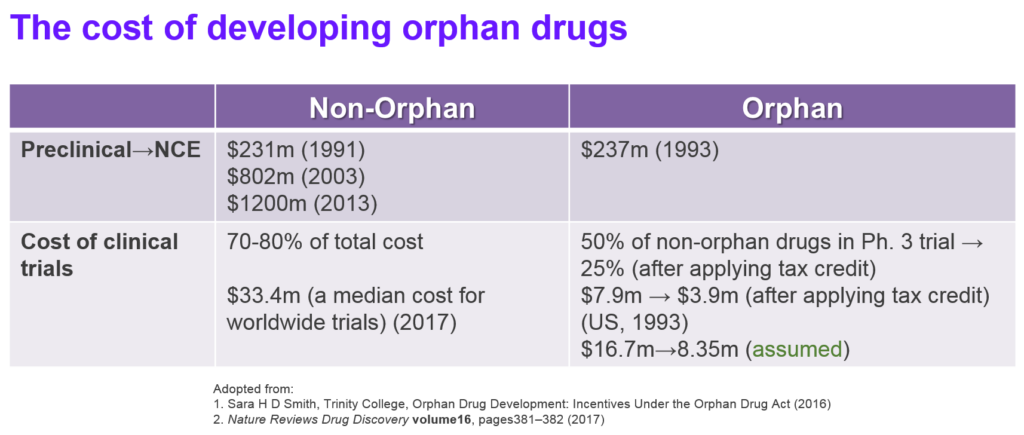
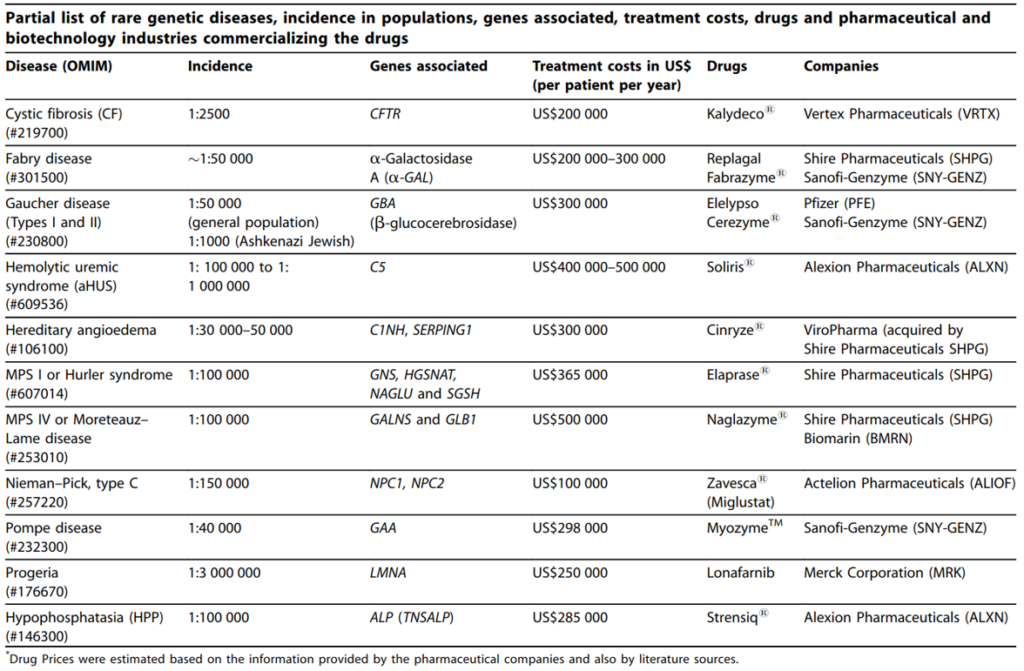
表3列舉了部分治療因基因異常導致的遺傳性罕見病的藥物治療成本(以每個病患每年的用藥費用為計算基礎),在美國的平均治療成本是147,308美元,而非罕見病患者的用藥成本是30,708美元5。雖然罕見病患者的數量有限,若將部分罕見病用藥的高藥價納入考量,這些藥物的仍能進入重磅藥物 (blockbuster)名單(年銷售額達成10億美元),例如由Alexion Pharmaceuticals開發及銷售的Soliris®。
罕見病藥物中的重磅藥物
全球罕見病藥物的年銷售額估計將從2018年的1,380億美元成長至2024年的2,620億美元(年複合成長率為11.38%)7,部分罕見病藥物甚至可以進入年銷售額達10億美元的重磅藥物名單,例如用於治療克隆氏症(Crohn’s disease)的Remicade®、治療多發性硬化症(multiple sclerosis)的Copaxone®以及用於治療非何杰金氏淋巴瘤(non-Hodgkin’s B-cell lymphoma)的Rituxan®,其中有部分藥物自首次上市距今已超過20年仍能高居重磅藥物之位。如表4所示,三種重磅藥物中的Remicade®和Rituxan®也在中國上市。
| Drug name | Remicade® | Copaxone® | Rituxan® |
| First approved indication (US) | Crohn’s disease | multiple sclerosis | non-Hodgkin’s B-cell
lymphoma |
| Launched year (US) | 1998 | 1997 | 1994 |
| Sales in 2017 ($ M) | 7,755 | 3,801 | 7,504 |
| Launched in China, Year, Indication | Yes, 2007,
Crohn’s disease |
No | Yes, 2002, Follicle center lymphoma |
另外,根據 Drugs to Watch 2018 的統計,羅氏最近剛被FDA批准上市、用於治療A型血友病的Hemlibra®,在2019年將可達到重磅級別,預計年銷售額達14.57億美元。另外,用於治療遺傳性家族性轉甲狀腺素類澱粉蛋白變性 (hereditary transthyretin-mediated amyloidosis, hATTR amyloidosis)的Patisiran,是首個(first-in-class) RNA干擾(RNAi)藥物 ,剛於2018年8月由FDA批准上市,估計在2021年可達重磅級別,年銷售額11.04億美元。
武田製藥巨額收購罕見病藥物龍頭Shire
在2018年初,日本的武田製藥 (Takeda) 開價623億美元收購罕見病藥物龍頭 Shire。Shire 目前在罕見病共有33個已上市的藥物且其產品線上還有超過100個藥物在研。透過 Cortellis的虛擬併購 (virtual merger) 分析,在武田併購Shire後可以强化臨床後期的産品線,且合併後的公司將儕身全球前10大藥廠之列。
罕見病藥物在中國的可及性
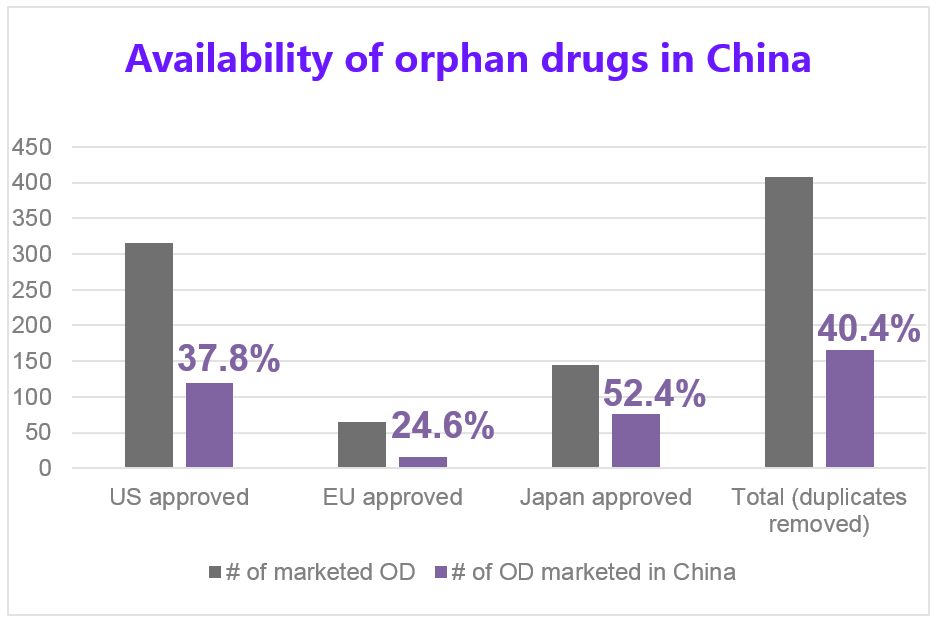
公布第一批罕見病名單開啓了在中國發展罕見病用藥的第一扇門,在中國取得已上市罕見病藥物也是在此領域發展的重要議題。根據Gong et al.在2012年的調查報告 8 中指出,在美國、日本、歐洲已核准的408個罕見病藥物中,僅有40.4% (408個罕見病藥物中的165個)可以在調查中的24間三甲醫院購得藥物(見圖6),其中18.9%是原廠藥,45.8%是學名藥,又165個中的120個罕見病藥物可由醫保給付;平均的治療費用為4,843.5美元,相當於中等收入的城市居民505.6天的收入。根據Gong的調查,大部分可取得的罕見病藥物仍是超過一般民眾的負荷。
發生在中國的主要罕見病包括苯酮尿症 (phenylketonuria), (thalassemia)、重型海洋性貧血 (thalassemia)、成骨不全症 (osteogenesis imperfecta)、高血氨症 (hyperammonemia)、有機酸血症 (organic academia) 以及威爾森氏症 (Wilson’s disease)、重症肌無力 (Myasthenia gravis) 、結節性硬化症 (tuberous sclerosis complex)、白化病(albinism)等。其他的罕見病如血友病 (hemophilia)、龐貝氏症 (Pompe disease) 和因醣脂質無法分解而堆積在體內造成的法布瑞氏症 (Fabry disease),其中在中國被診斷出法布瑞氏症的病患不到5%。發生在兒童的則有雄激素不敏感症 (androgen insensitivity syndrome)、肋膜肺母細胞瘤 (pleuropulmonary blastoma)、橫紋肌肉瘤(rhabdomyosarcoma,含腺泡型(alveolar)及胚胎型(embryonal)二種子型)、Blau症候群 (Blau syndrome)、Bardet-Biedl氏症候群(Bardet-Biedl Syndrome) 以及先天性眼球震顫(Infantile nystagmus syndrome)…等兒科罕見病。
罕見病或法律定義上的孤兒疾病 (orphan disease) 約影響中國1600萬人,估計的發病率約是每30-50萬人約有1名病患,目前在中國的公布罕見病數據尚不完整,也沒有足够的財務支援罕見病治療,其中僅有部分的患者能得到治療。目前中國國家藥品監督管理局 (NMPA) 尚未公布關於罕見病在中國的特別法及相應條款,使得罕見病患者接受治療的可及性受到影響。
多數情况下,主要藥政市場的監管單位界定罕見病會用兩個標準,一個是絕對數量,比如低於25萬人口,就可以定義罕見病。一個是百分比,比如低於萬分之一等,做為罕見病的基礎定義。
然而,中國有著13億的人口基數,這二個標準,無論用哪個,將都是一個值得關注的數字,這會對支付部門(醫保或者保險公司)帶來巨大負擔。第一批罕見病目錄的121罕見病,它既不看發病率患病率,也不看絕對數字,而是建設性給出一個目錄,相信是徵求了臨床醫師需求意見後給出的。考量中國自身的首要需求,率先推進一批罕見病的研發和治療。
非腫瘤罕見病用藥在中國的臨床試驗趨勢
觀察第一批罕見病目錄的121個罕見病是屬於非腫瘤的罕見病,請參考圖7和圖8,進一步分析近10年在中國的臨床試驗及非腫瘤罕見病用藥的臨床試驗趨勢。平均而言,在中國的執行的臨床試驗中,4.3%爲非腫瘤罕見病的臨床試驗。排除臨床試驗期別無法確定的數量後,當中又以臨床二期(通常被視為樞紐試驗 pivotal trials)的數量最多,緊接著的是臨床四期(上市後的藥品監控試驗)以及臨床三期的試驗;在中國執行的多爲後期的臨床試驗,估計和上市策略或與監管法規要求有關。
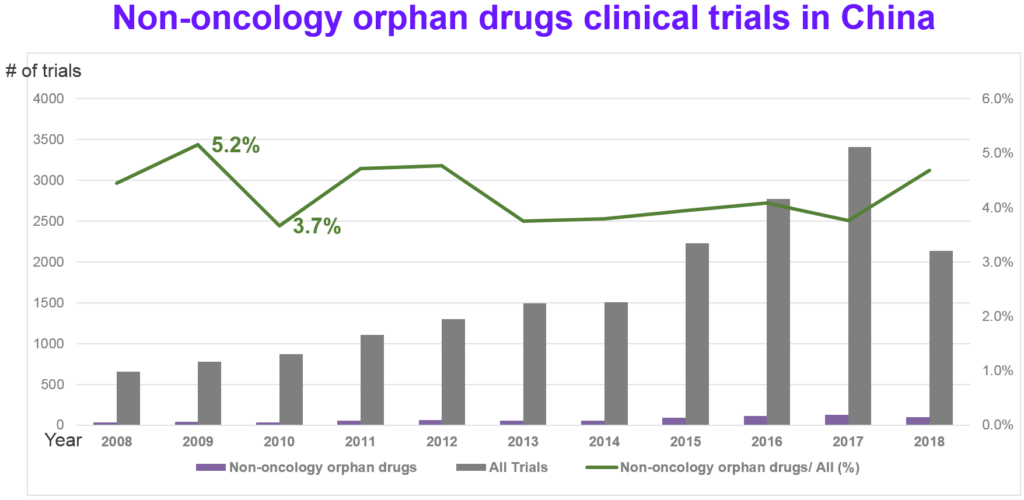
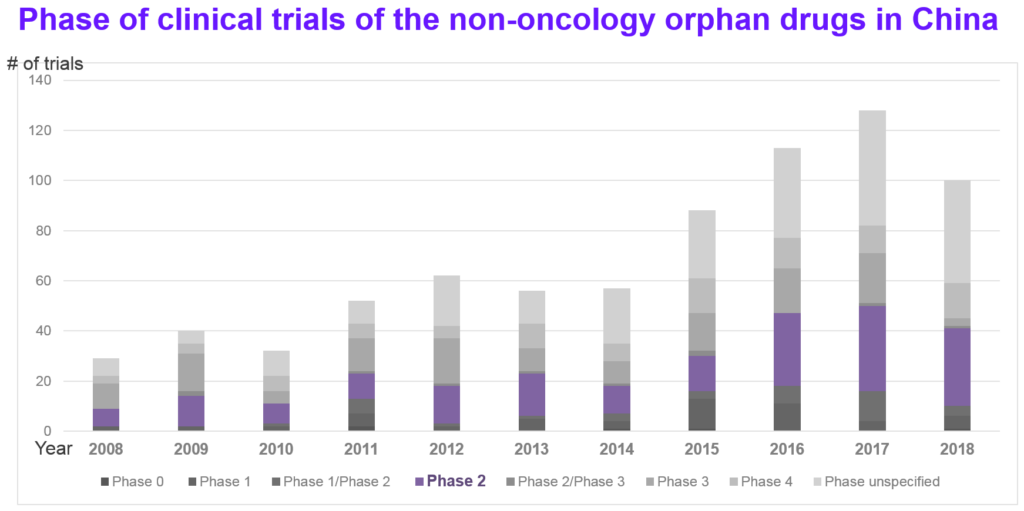
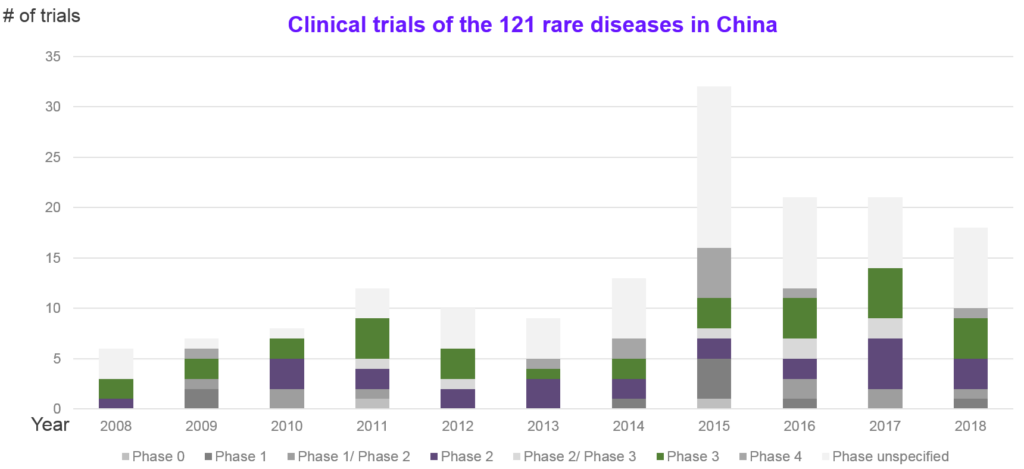
整體而言,2015年後公開的臨床試驗數量有明顯成長,如圖9所示,第一批罕見病名單的121個適應症在中國的臨床試驗,也是後期的臨床試驗的數量最多。
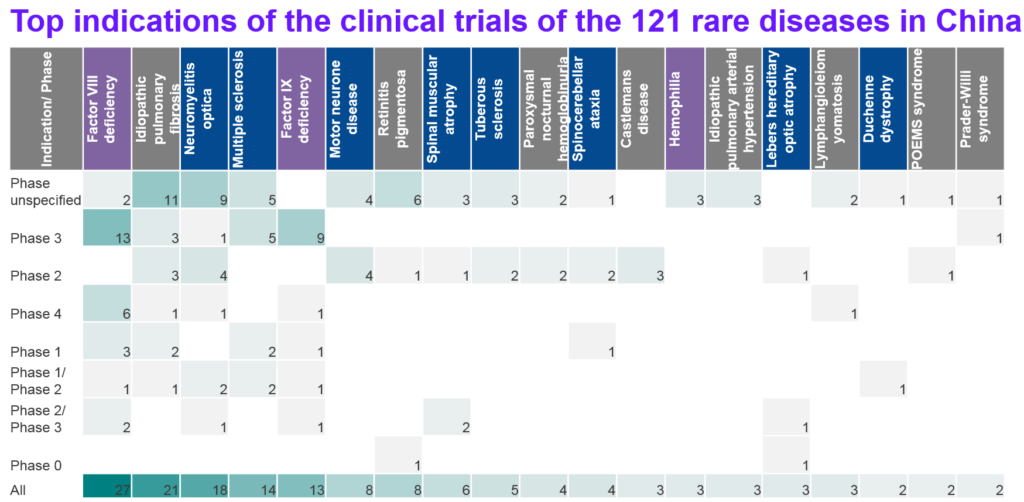
進一步分析第一批罕見病名單中的臨床試驗,如圖10所示,比例最高的是神經疾病相關的罕見病,佔40%,其次是血液相關的罕見病(佔29%),包括第八凝血因子缺乏 (factor VIII deficiency)、第九凝血因子缺乏 (Factor IX deficiency)以及血友病,與前述非腫瘤罕見病的臨床試驗相似,也是後期的臨床試驗居多數。
中國自2015年開始進行藥政改革,目的是鼓勵創新及减少監管法規所帶來的負擔,新的罕見病藥物可適用包括優先審評、接受境外臨床試驗數據以及臨床試驗改採備案制等措施,在在反應出NMPA對創新及加速新藥上市的重視。預期未來中國正式公佈罕見病的定義後,對於在中國發展罕見病的遠景將會更清晰。
罕見病藥物在中國的願景
根據Gong et al.的研究,在美國、歐洲和日本核准上市的罕見病藥物中,分別僅有37.8%、24.6%和52.4%能在中國的三甲醫院購藥。相較於上述國家,中國尚未公布依據人口或患病率而定的罕見病的官方定義,也還沒公布針對罕見病上市的特別法或優惠。NMPA可依申請人的請求豁免臨床試驗或减少受試者人數;在其他國家已核准上市的罕見病藥物,NMPA可能考慮有條件的批准上市,並要求申請人在上市後補充試驗數據。
就現有的罕見病名單來看,身為全球第二大醫藥市場的中國,目標是加速罕見病的診斷及新藥上市;罕見病名單的公布對於後續的政策方向提供醫療照護、財務支持的基礎。中國政府也重視建立非盈利的罕見病研究機構,例如:中國罕見病發展中心 (Chinese Organization for Rare Disorders, CORD) 成立於2013年,是一家專注於罕見病領域的非營利組織。罕見病在中國的發展可以預期的是若由國家計劃保證覆蓋明確的罕見病,將推動中國罕見病患者的醫療保障照顧,對於已在其他國家上市的罕見病藥物, NMPA可能以特別法規範其上市及銷售,使得病患能得到妥善治療及减少監管冗餘及經濟上的負擔。其他的關鍵措施可能還包括:比照其他藥政市場允許罕見病以快速通道的上市路徑、免除大規模臨床試驗、對罕見病藥物的特別許可以及配合建立病患網絡和治療費用報銷等方式。 上述種種將使中國在罕見病的醫療照護和社會負擔上,位於一個有利位置。
參考資料:
1. GlobalGenes.org
2. GlobalGenes.org
3. Sara H D Smith, Trinity College, Orphan Drug Development: Incentives Under the Orphan Drug Act (2016)
4. Nature Reviews Drug Discovery volume 16, pages 381–382 (2017)
5. Drug Discovery Today. 2018 Jan;23 (1):187-195)
6. Drug Discovery Today. 2018 Jan;23 (1):187-195)
7. Evaluate Pharma, Orphan Drug report (2018))
8. Gong et al. Orphanet Journal of Rare Diseases (2016) 11:20


